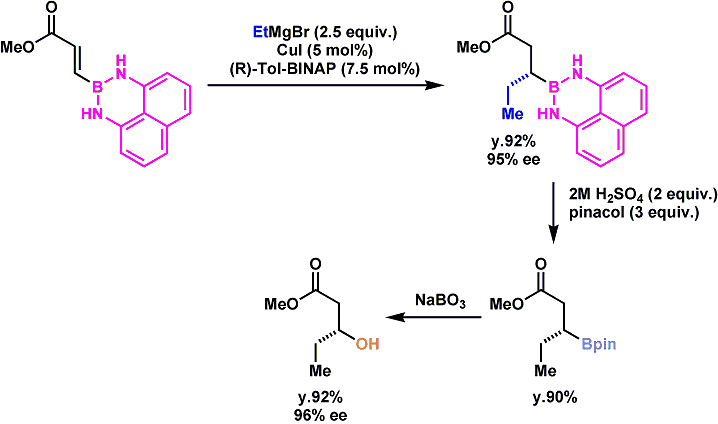
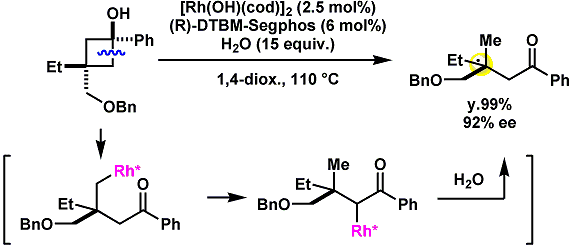
10.1002/anie.201000472
クロスカップリングの基質としては通常アリールハライド、トリフレートなどが用いられるが、反応性の高さと対応するアニリンから容易に調製することが可能ということを考えるとジアゾニウム塩の利用は一つの魅力的な代替案になりうる。実際、アレーンジアゾニウム塩はHeck反応や鈴木カップリング、Stilleカップリングなどのパラジウム触媒反応には適応されてきている。本報告はこのジアゾニウム塩の化学を薗頭反応に拡張しようというものだ。
筆者らはまず各種パラジウム触媒、塩基、溶媒にて反応を試したが目的とするアリールアルキンは得られず、複雑な混合物を与えることが多かった。そこで、ヨウ化物イオンを系中に添加することでヨードデジアゾニウム化を起こすことを試みることとした。すると2当量のTBAI添加により良好な収率で目的物が得られることがわかった。
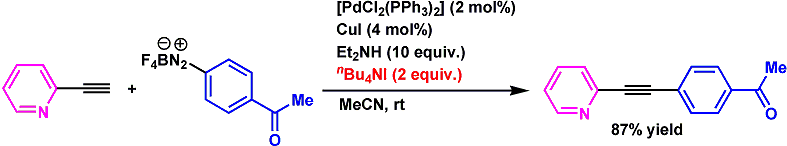
最適条件下、基質一般性の検討を行ったところ、電子供与基、吸引基いずれを有するアリールジアゾニウム塩も良好に反応することがわかった。またアルキン部位の一般性も直鎖アルキル基から2-ピリジルのような触媒毒となりうる置換基でも良好な収率で目的物を得ることに成功している。さらに一端ジアゾニウム塩を単離することなしに、アニリンからワンポットにてアリールアルキンまで高収率で導くことにも成功している。
興味深いのはTBAIを添加することで何が起きているのかである。筆者らは対象実験の結果から、まず脱ジアゾニウムによるヨウ化アリールの生成がおき、続いてパラジウム触媒による酸化的付加がおきて反応が進行していると述べている。別途調製した[PhPdI(Ph3P)2]、[PhPdBr(Ph3P)2]をアルキンと反応させたところ臭化物イオン由来の化学種の方が高活性であったことと、ヨウ化物イオンの変わりに臭化物イオン源としてTBABを用いた場合には目的物は得られずブロモベンゼンが高収率で得られたことから、この場合はブロモベンゼンへの酸化的付加が律速段階になっているということだろうか。
収率、官能基受容性ともに高く魅力的な反応と言えるけれど、系中でヨウ化アリールを発生させているなら必ずしもジアゾニウム塩を経由する必要がないとも言えるのがアピールに弱い気がする。
参考)以前紹介したフェノール塩を基質とするクロスカップリング反応